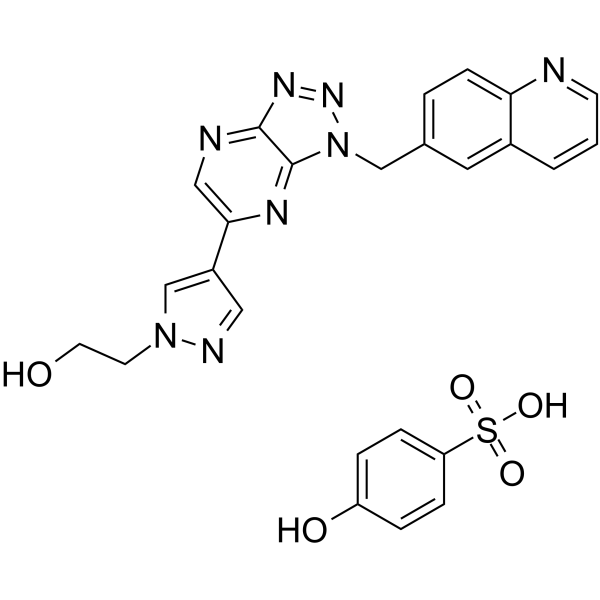
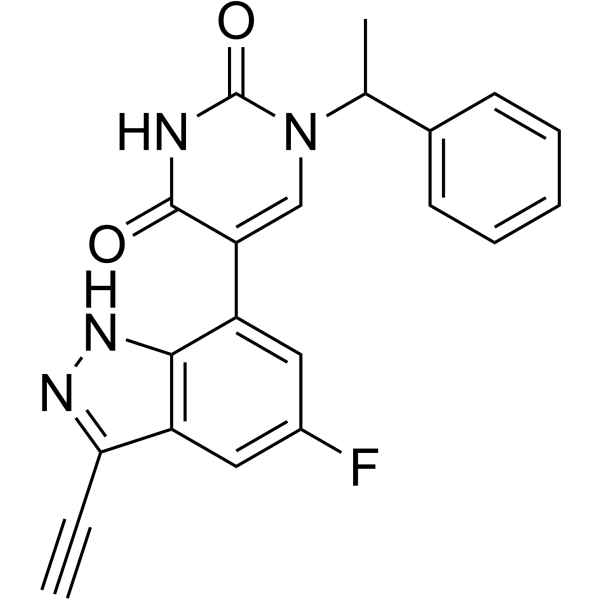
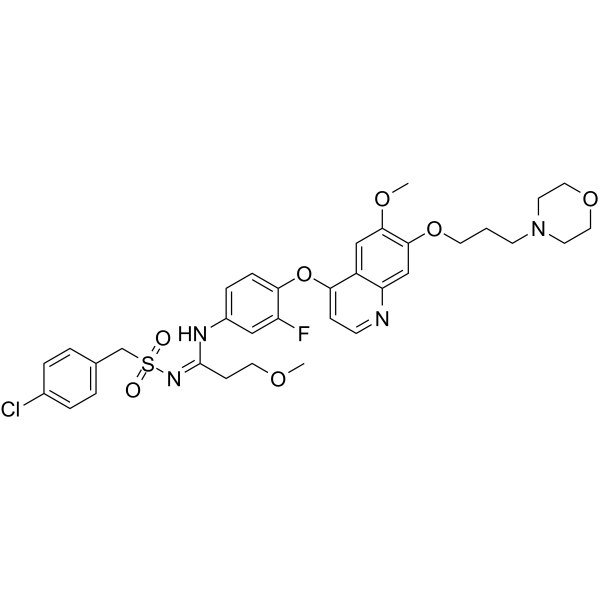
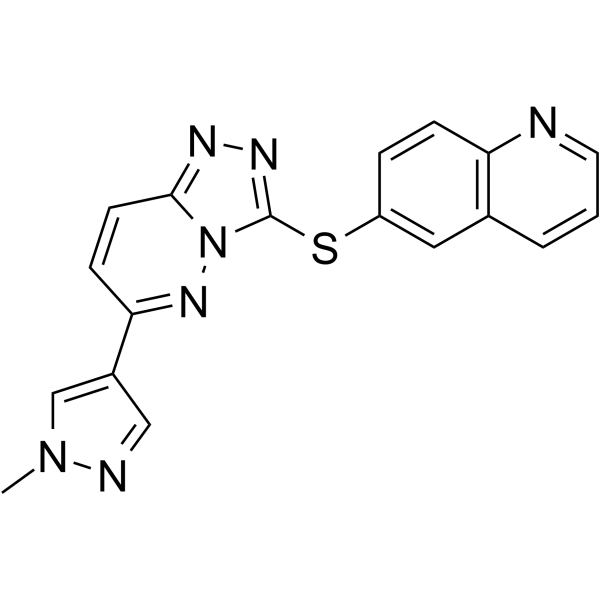
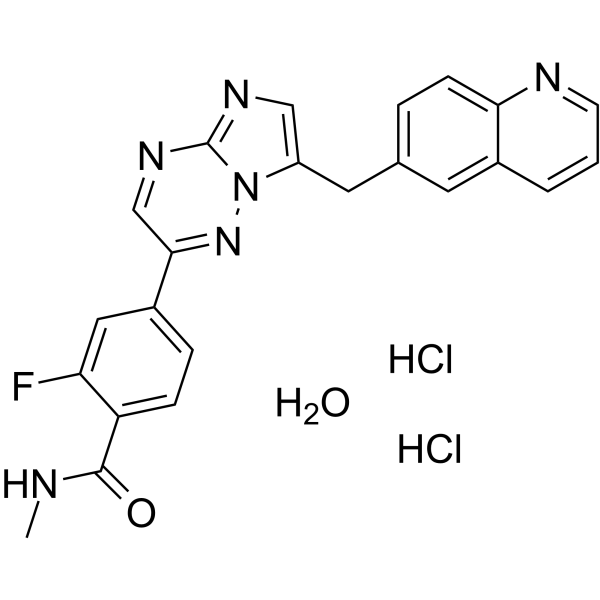
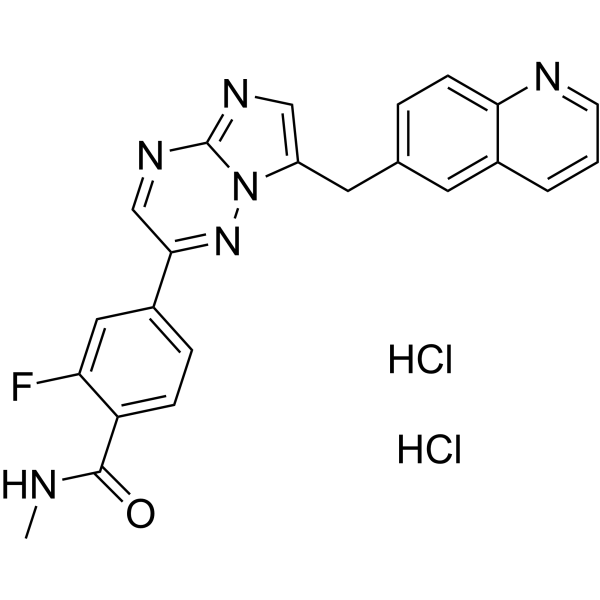
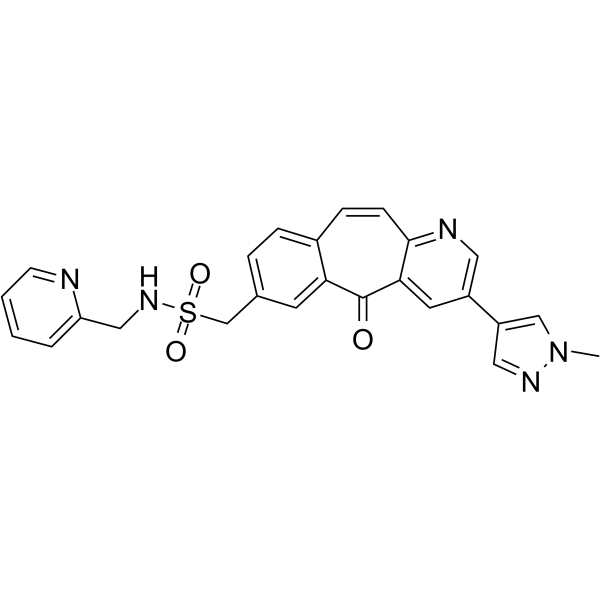
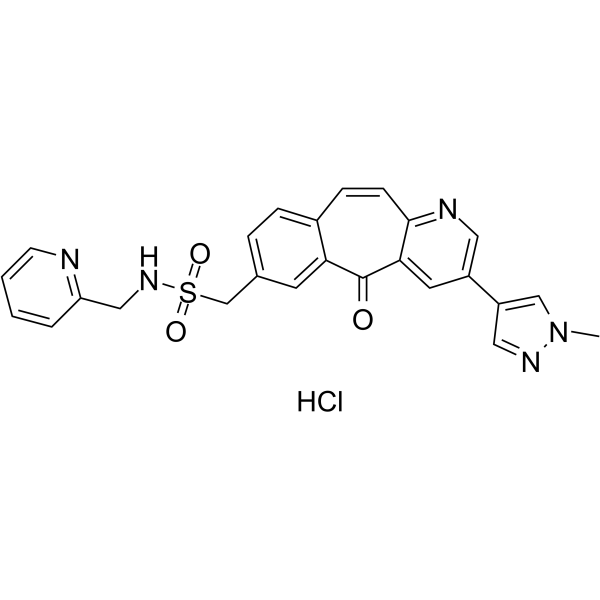
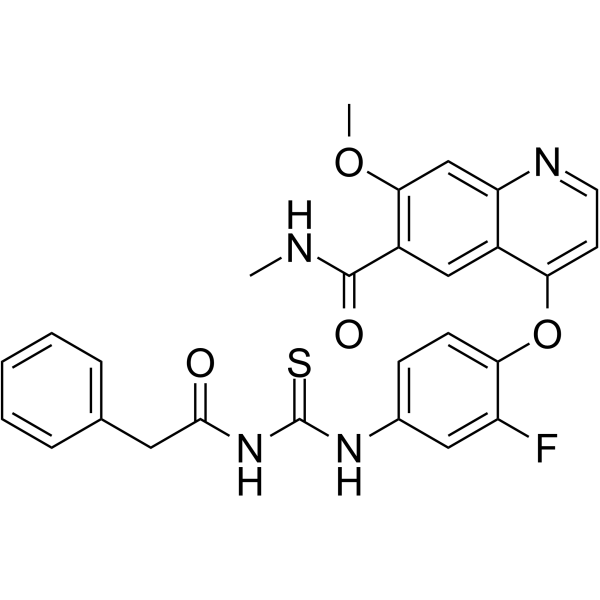
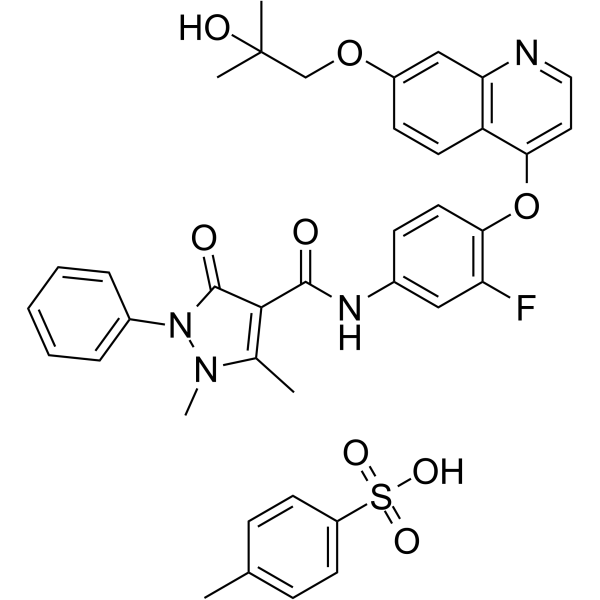
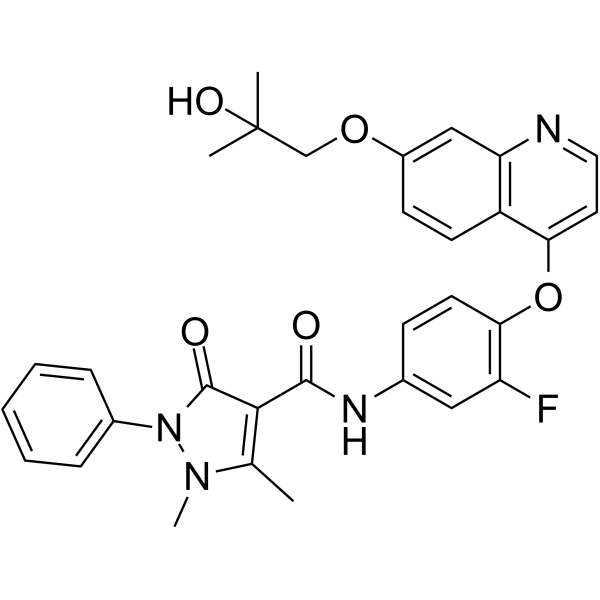
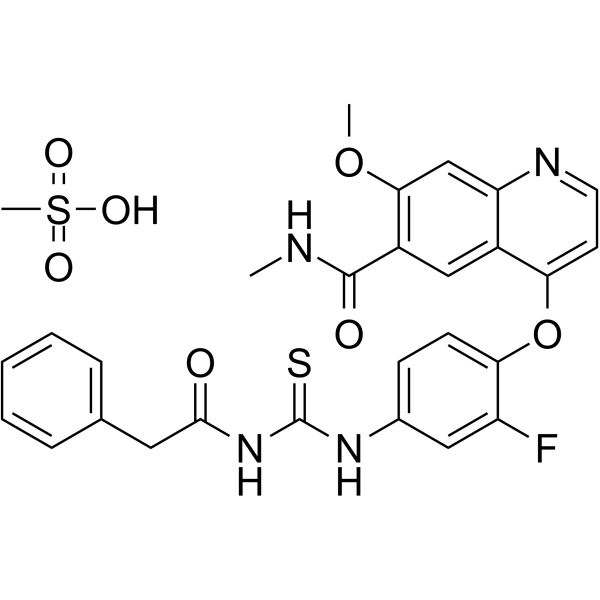
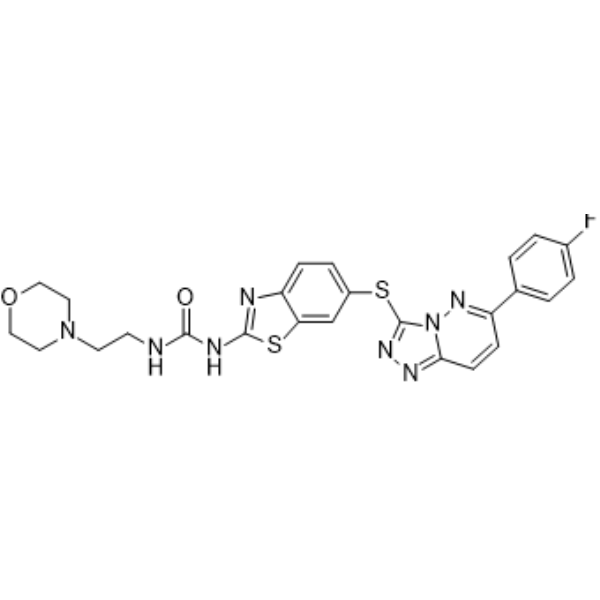
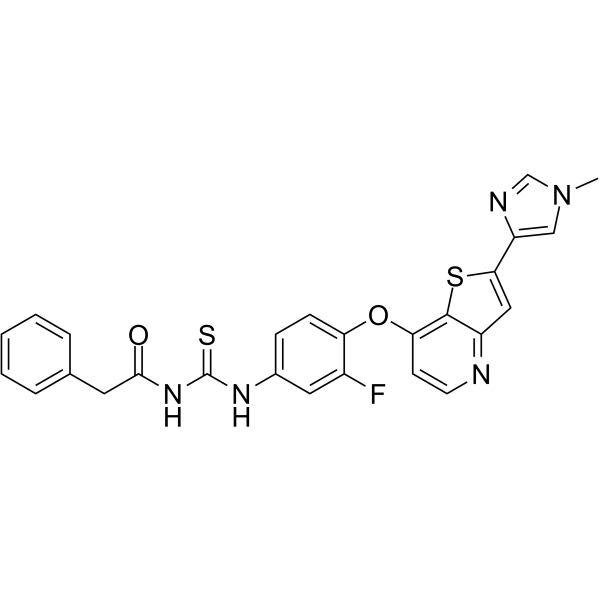
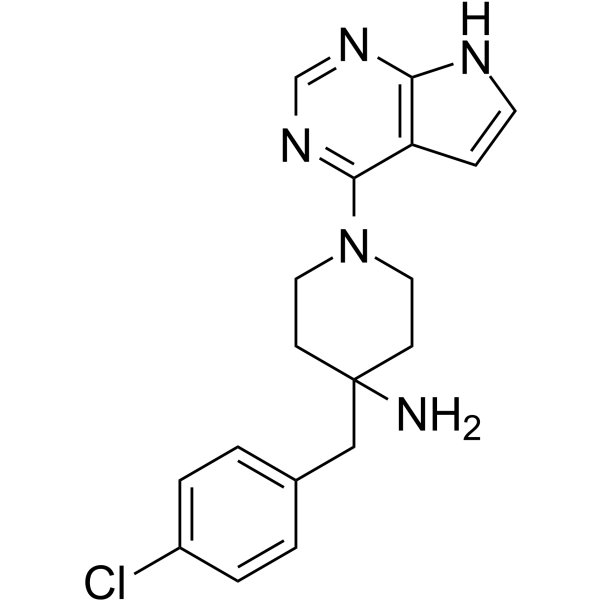
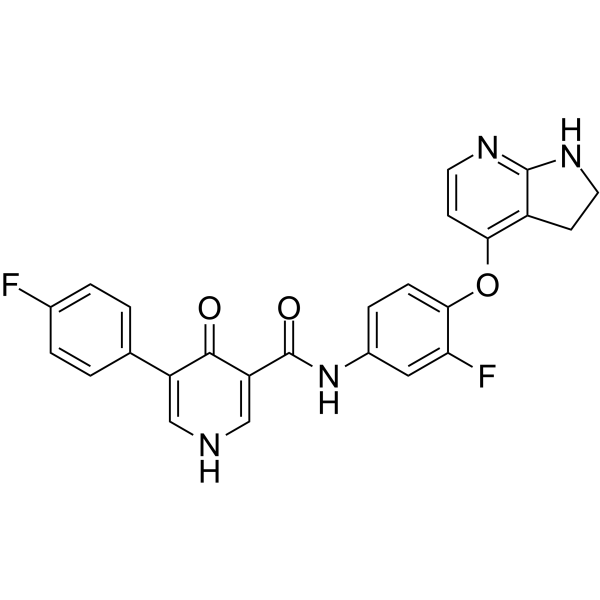
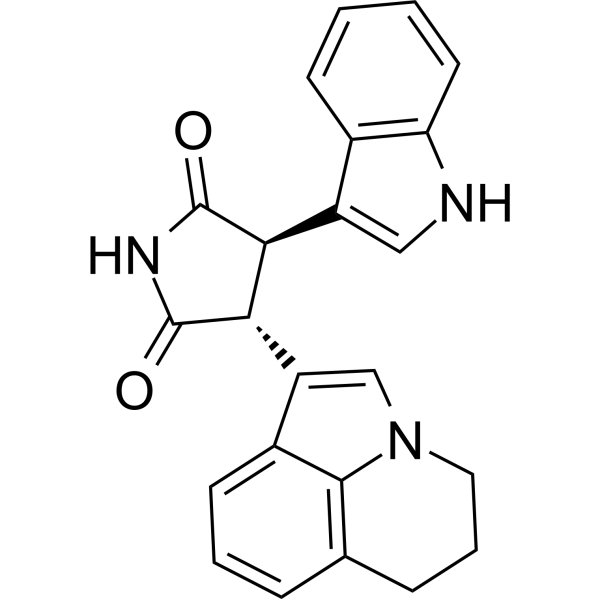
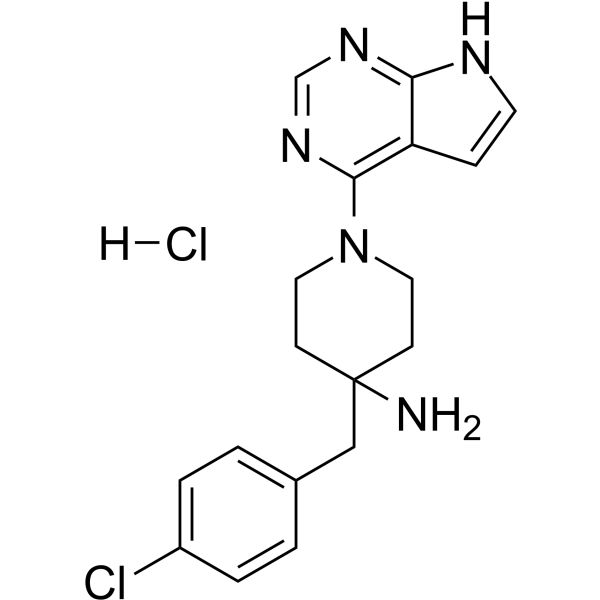
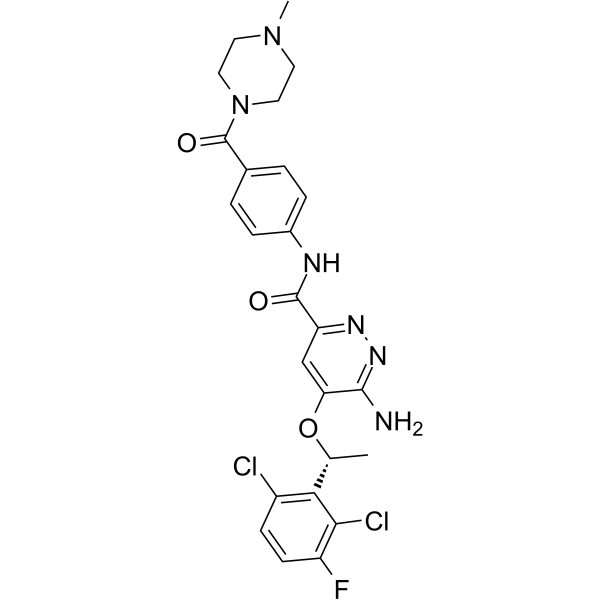
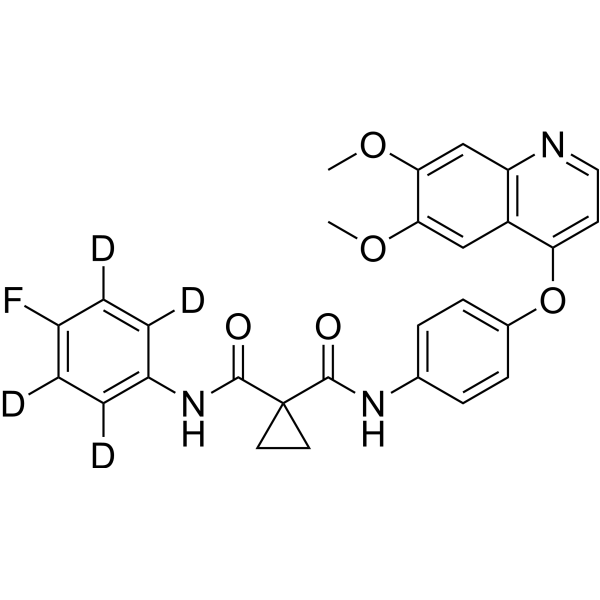
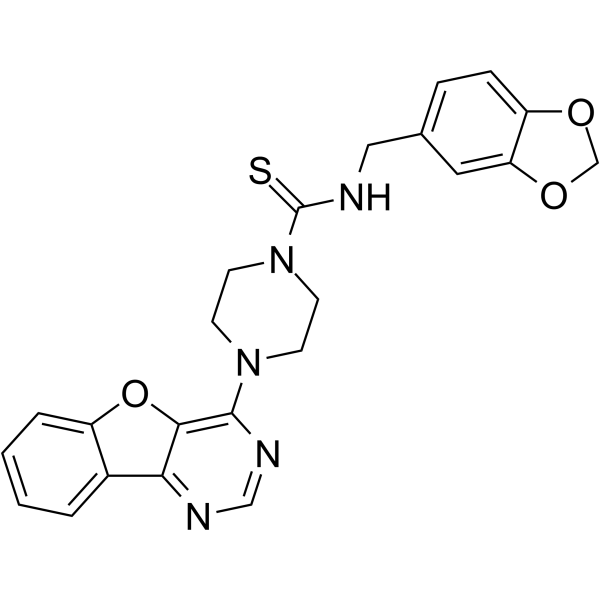
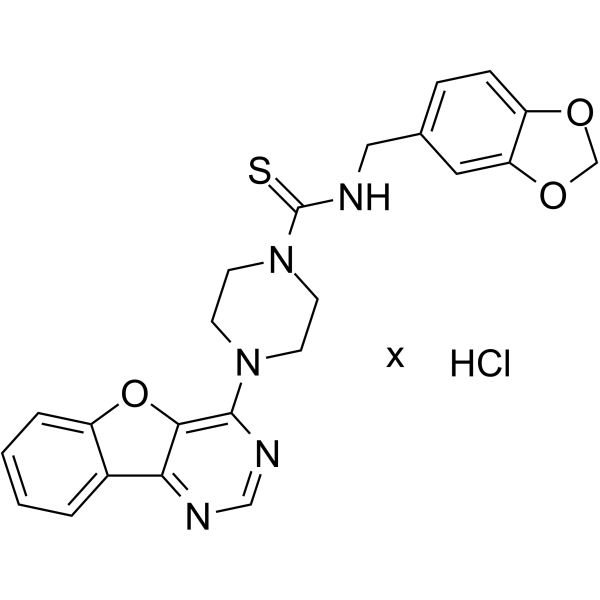
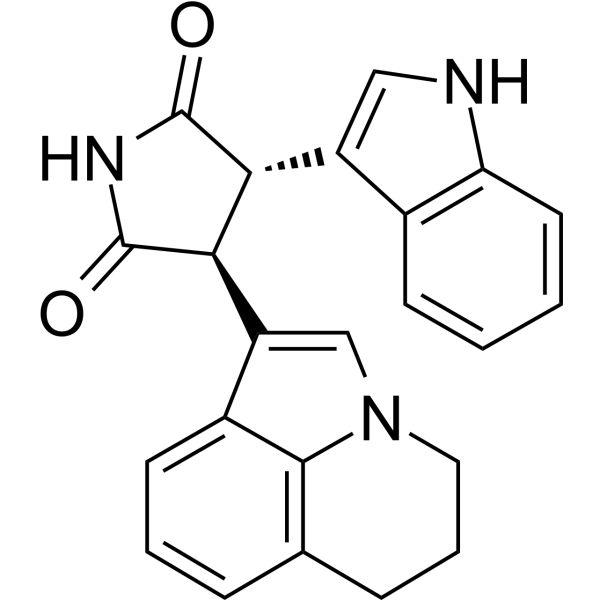
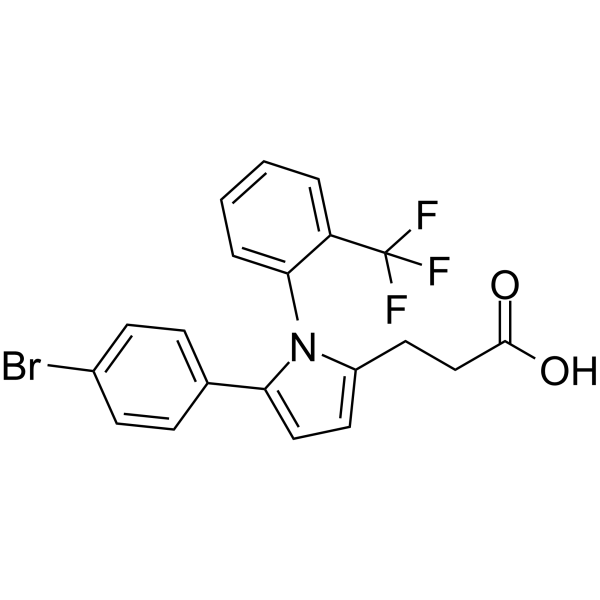
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
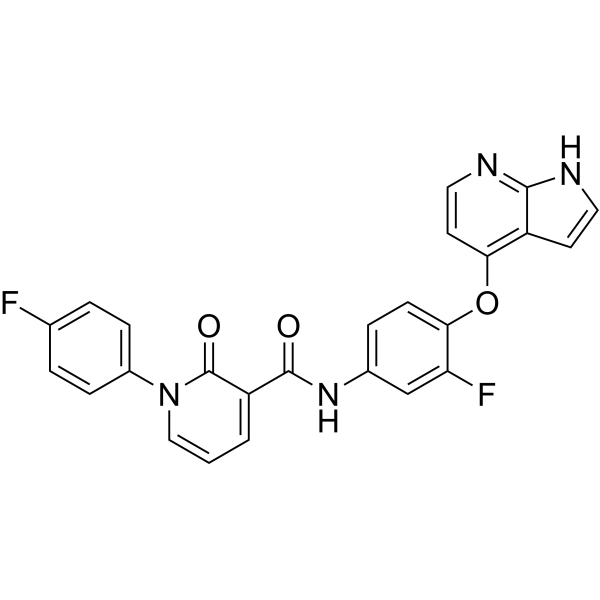
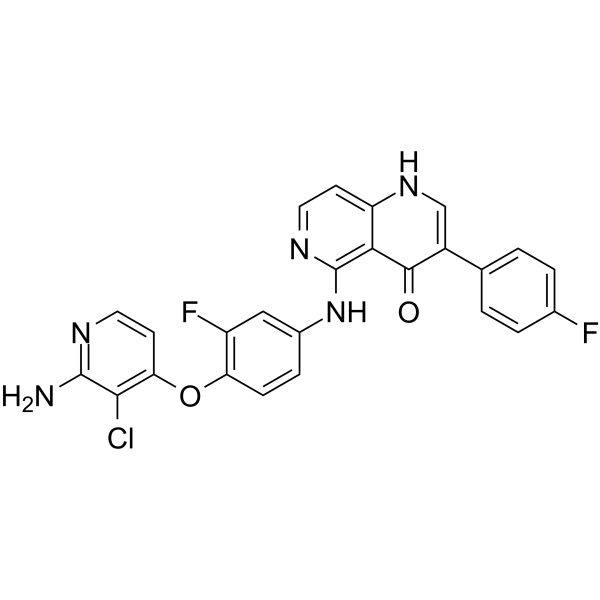
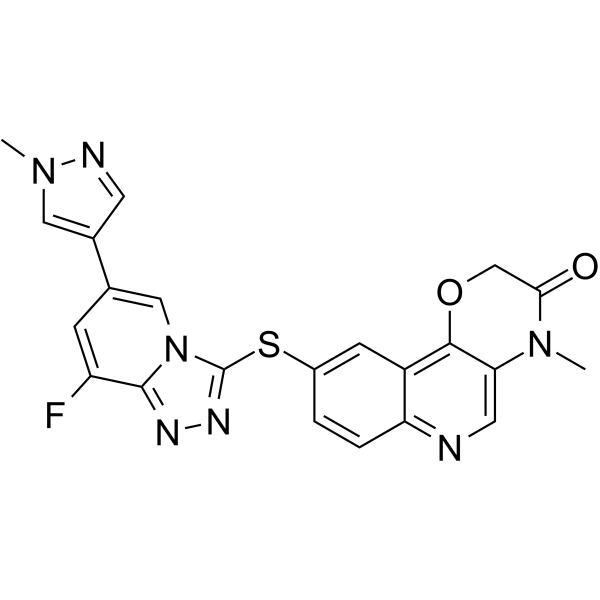
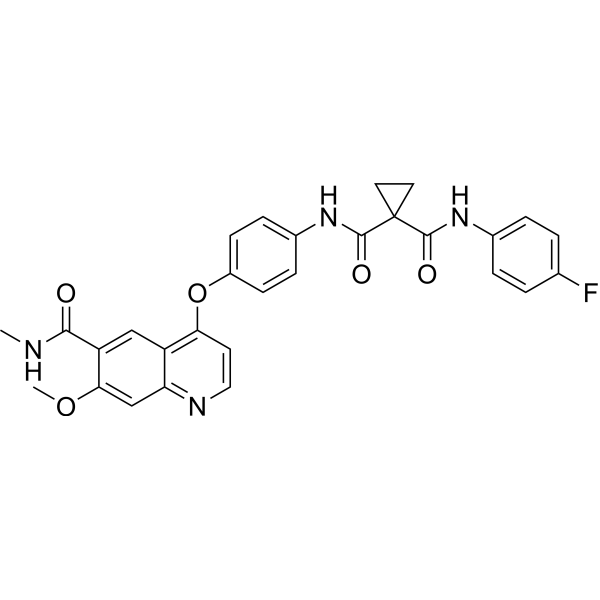
METkinase-IN-4 is an orally active Metkinase inhibitor. METkinase-IN-4 has potent Metkinase inhibitory activity with an IC50 value of 1.9 nM. METkinase-IN-4 can be used for the research of cancer .
DS-1205b free base is a potent and selective inhibitor of AXL kinase, with an IC50 of 1.3 nM. DS-1205b free base also inhibits MER, MET, and TRKA, with IC50s of 63, 104, and 407 nM, respectively. DS-1205b free base can inhibit cell migration in vitro and tumor growth in vivo .
METkinase-IN-3 (compound 8) is an orally active and potent MET inhibitor, with an IC50 of 9.8 nM. METkinase-IN-3 shows good and broad-spectrum antiproliferative activity against cancer cell lines .
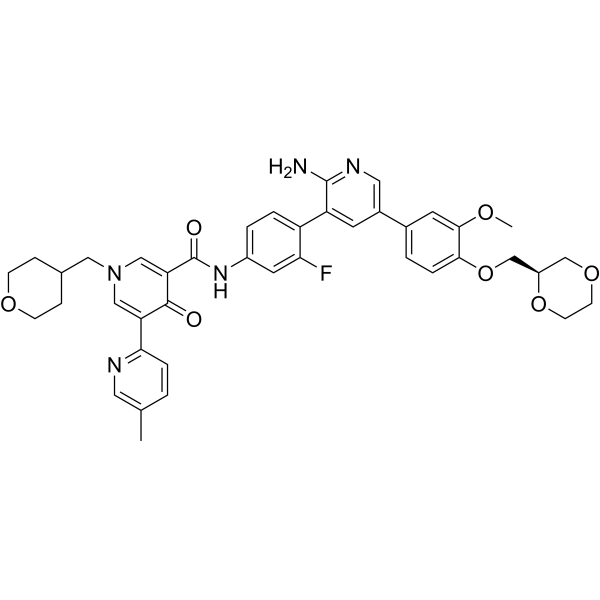
c-Met-IN-12 (compound 4r) is an orally active, potent and selective type II c-Metkinase inhibitor, with an IC50 of 10.6 nM. c-Met-IN-12 displays high inhibitory effects (inhibition rate > 80% in 1 μM) against AXL, Mer and TYRO3kinases. c-Met-IN-12 can be used a scaffold for further kinase selectivity enhancement. c-Met-IN-12 shows antitumor efficacy .
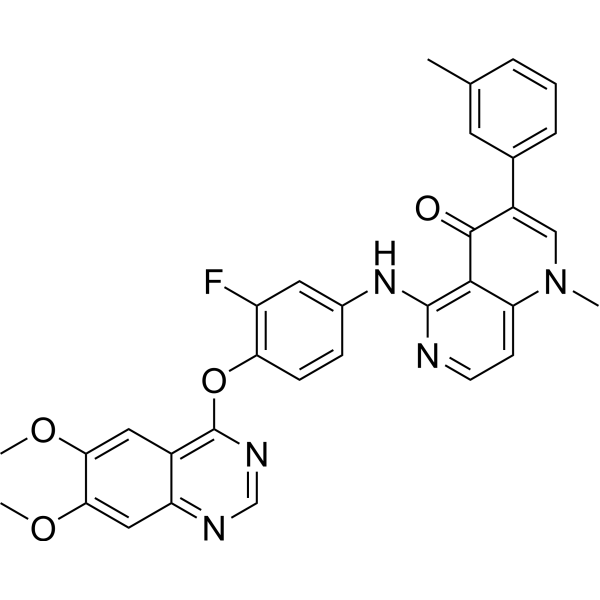
Zgwatinib (SOMG-833) is a potent, selective, and ATP-competitive c-MET inhibitor, with an IC50 of 0.93 nM against c-MET, over 10,000-fold more potent compared with 19 tyrosine kinases (including c-MET family members and highly homologous kinases). Zgwatinib potently inhibits c-MET-driven cell proliferation. Zgwatinib as a potential candidate agent for c-MET-driven human cancers research .
JNJ-38877618 is a potent, highly selective, orally bioavailable Metkinase inhibitor with IC50s of 2 and 3 nM for wild type and mutant Met, respectively.
PF-04217903 is a potent ATP-competitive c-Metkinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
PF-04217903 mesylate is a potent ATP-competitive c-Metkinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 mesylate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
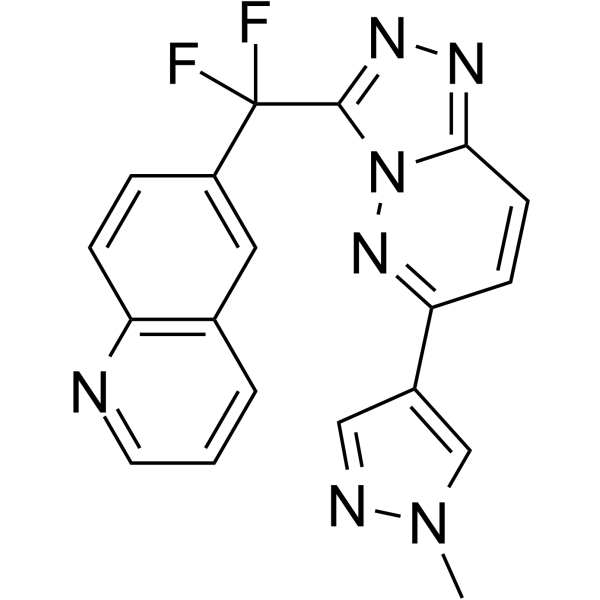
AMG-337 is a potent, orally active, selective METkinase inhibitor with IC50 values of 1, 1, 4.7, 5, 21.5, 1077 and >4000 nM of WT MET, H1094R MET, M1250T MET, HGF-stimulated pMET (PC3 cells) MET, V1092I MET, Y1230H MET, and D1228H MET, respectively. AMG 337 inhibits the phosphorylation of MET and downstream effectors in MET-amplified cancer cell lines, resulting in an inhibition of MET-dependent cell proliferation and induction of apoptosis .
Glumetinib (SCC244) is a highly selective, orally bioavailable, ATP-competitive c-Met inhibitor with an IC50 of 0.42 nM. Glumetinib has greater than 2400-fold selectivity for c-Met over those 312 kinases evaluated, including the c-Met family member RON and highly homologous kinases Axl, Mer, TyrO3. Antitumor activity .
TAM&Met-IN-1 is a potent TAM and c-Metkinase inhibitor with IC50 values of 6.1 nM, 13.2 nM and 21.6 nM for AXL, MER and TYRO3, respectively. TAM&Met-IN-1 can be used for researching anticancer .
PF-04217903 phenolsulfonate is a potent ATP-competitive c-Metkinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 phenolsulfonate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
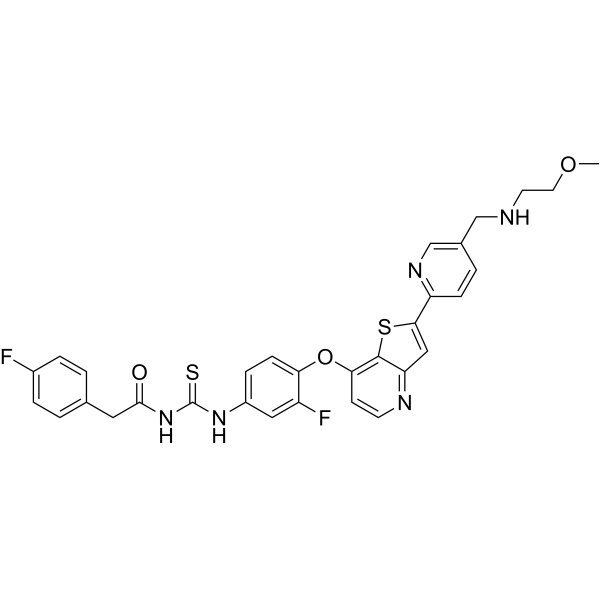
c-Met-IN-17 is a potent c-Metkinase inhibitor with an IC50 of 0.031 μM. c-Met-IN-17 can be used in anticancer research. . c-Met-IN-17 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
c-Met-IN-14 (compound 26af) is a selective inhibitor of c-Metkinase from N-sulfonylamidine-based derivatives, with an IC50 value of 2.89 nM. c-Met-IN-14 shows anticancer activity by blocking phosphorylation of c-Met, and arrests cell cycle at G2/M phase. c-Met-IN-14 induces apoptosis of A549 cells in a dose-dependent manner .
SGX523 is a exquisitely selective and ATP-competitive MET inhibitor. SGX523 potently inhibits MET with an IC50 of 4 nM and is >1,000-fold selective versus other protein kinases. Antitumor activity .
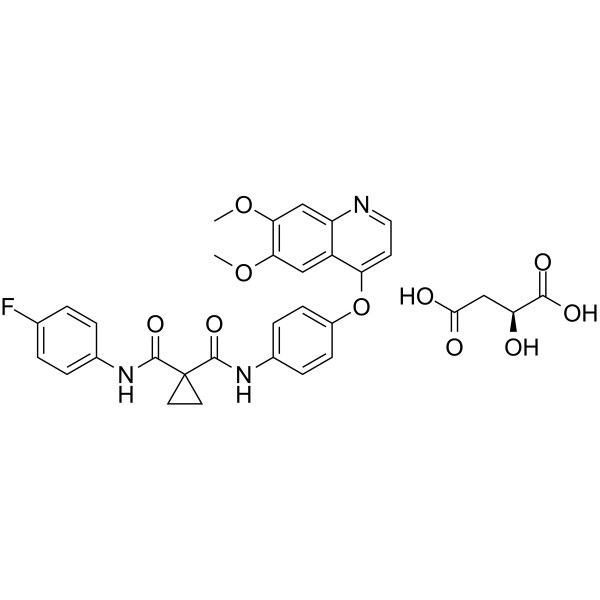
Dalmelitinib is an orally active selective c-Metkinase inhibitor (IC50: 2.9 nM) that binds to the ATP-binding region of c-Met. Dalmelitinib induces the phosphorylation of MET, partially or completely inhibits the phosphorylation of AKT and ERK. Dalmelitinib potently inhibits cancer cell (c-Met oncogene amplification) proliferation, and is used for the research of cancers like human non-small cell lung cancer (NSCLC) .
JNJ-38877605 is an orally active ATP-competitive inhibitor of c-Met with an IC50 of 4 nM, 600-fold selective for c-Met than 200 other tyrosine and serine-threonine kinases . JNJ-38877605 inhibits c-Met phosphorylation and regulates lipid accumulation. JNJ-38877605 can be used for tumor and metabolic disease reseach .
PHA-665752 is a selective, ATP-competitive, and active-site inhibitor of the catalytic activity of c-Metkinase (Ki=4 nM; IC50=9 nM). PHA-665752 exhibits >50-fold selectivity for c-Met compared with a panel of diverse tyrosine and serine-threonine kinases. PHA-665752 induces apoptosis and cell cycle arrest, and exhibits cytoreductive antitumor activity .
(Rac)-Tivantinib is the isomer of Tivantinib (HY-50686), and can be used as an experimental control. Tivantinib is a highly selective c-Met tyrosine kinase inhibitor with a Ki of 355 nM.
c-Met-IN-10 (compound 26a) is a highly potent c-Metkinase inhibitor with an IC50 value of 16 nM. c-Met-IN-10 has inhibitory activity against cancer cells A549, H460 and HT-29 with IC50s of 0.56 ~ 1.59 μM. c-Met-IN-10 suppresses the colony formation on HT-29 cells, induces HT-29 and A549 cells apoptosis, and inhibits A549 cells motility. c-Met-IN-10 can be used for researching anticancer .
BMS 777607 (BMS 817378) is a Met-related inhibitor for c-Met, Axl, Ron and Tyro3 with IC50s of 3.9 nM, 1.1 nM, 1.8 nM and 4.3 nM, respectively, and 40-fold more selective for Met-related targets than Lck, VEGFR-2, and TrkA/B, with more than 500-fold greater selectivity versus all other receptor and non receptor kinases .
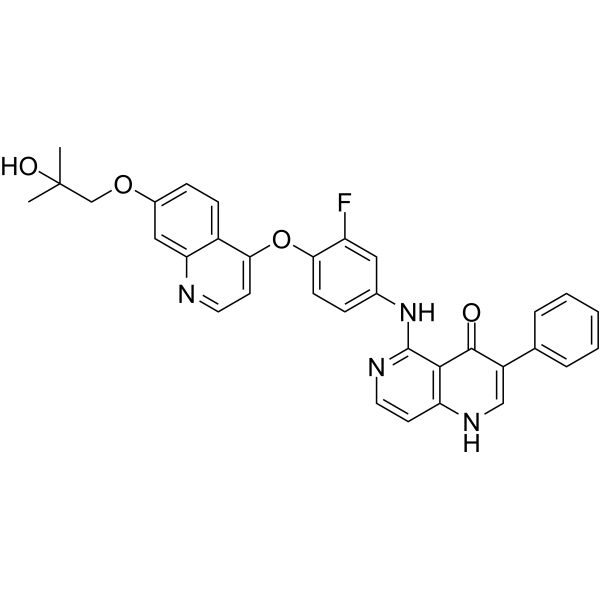
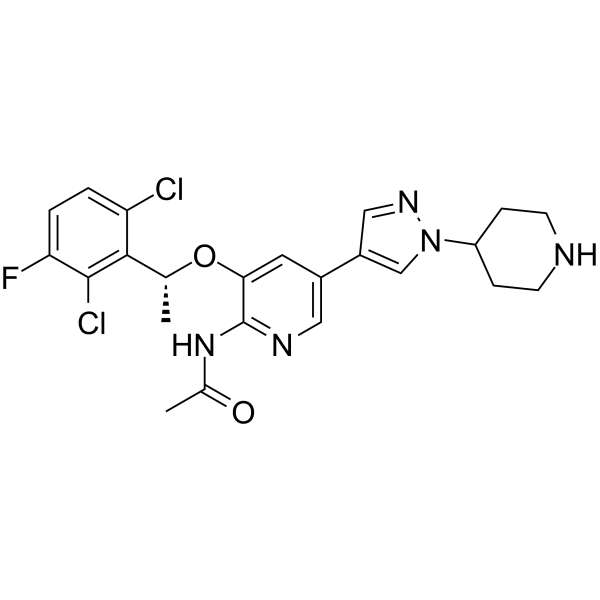
Bozitinib (PLB-1001) is a highly selective c-METkinase inhibitor with blood-brain barrier permeability. Bozitinib (PLB-1001) is a ATP-competitive small-molecule inhibitor, binds to the conventional ATP-binding pocket of the tyrosine kinase superfamily .
Capmatinib (INC280; INCB28060) is a potent, orally active, selective, and ATP competitive c-Metkinase inhibitor (IC50=0.13 nM). Capmatinib can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) hydrochloride is a potent, orally active, selective, and ATP competitive c-Metkinase inhibitor (IC50=0.13 nM). Capmatinib hydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib hydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib hydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) dihydrochloride hydrate is a potent, orally active, selective, and ATP competitive c-Metkinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride hydrate can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride hydrate potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride hydrate is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) dihydrochloride is a potent, orally active, selective, and ATP competitive c-Metkinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
MK-8033 is an orally active ATP competitive c-Met/Ron dual inhibitor (IC50s: 1 nM (c-Met),7 nM (Ron)), with preferential binding to the activated kinase conformation. MK-8033 can be used in the research of cancers, such as breast and bladder cancers, non-small cell lung cancers (NSCLCs) .
MK-8033 hydrochloride is an orally active ATP competitive c-Met/Ron dual inhibitor (IC50s: 1 nM (c-Met),7 nM (Ron)), with preferential binding to the activated kinase conformation. MK-8033 hydrochloride can be used in the research of cancers, such as breast and bladder cancers, non-small cell lung cancers (NSCLCs) .
Pamufetinib (TAS-115) is a potent VEGFR and hepatocyte growth factor receptor (c-Met/HGFR)-targetedkinase inhibitor with IC50s of 30 and 32 nM for rVEGFR2 and rMET, respectively.
Ningetinib Tosylate is a potent, orally bioavailable small molecule tyrosine kinase inhibitor (TKI) with IC50s of 6.7, 1.9 and <1.0 nM for c-Met, VEGFR2 and Axl, respectively.
Ningetinib is a potent, orally bioavailable small molecule tyrosine kinase inhibitor (TKI) with IC50s of 6.7, 1.9 and <1.0 nM for c-Met, VEGFR2 and Axl, respectively.
Pamufetinib (TAS-115) mesylate is a potent VEGFRand hepatocyte growth factor receptor (c-Met/HGFR)-targetedkinase inhibitor, with IC50s of 30 and 32 nM for rVEGFR2 and rMET, respectively.
SAR125844 is a potent, selective, and ATP-competitive METkinase inhibitor with the value of IC50 is 4.2 nM and Ki is 2.8 nM. SAR125844 has antitumor activity and can be used for the research of cancer .
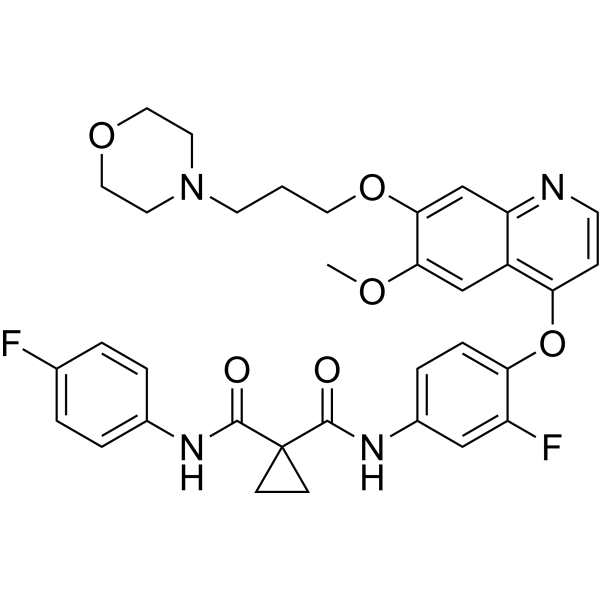
MGCD-265 analog is a potent and oral active inhibitor of c-Met and VEGFR2 tyrosine kinases, with IC50s of 29 nM and 10 nM, respectively. MGCD-265 analog has significant antitumor activity .
CCT128930 is a ATP-competitive and selective inhibitor of AKT (IC50=6 nM for AKT2). CCT128930 has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). Antitumor activity.
BPI-9016M is a potent, orally active, and selective dual c-Met and AXL tyrosine kinases inhibitor. BPI-9016M suppresses tumor cell growth, migration and invasion of lung adenocarcinoma .
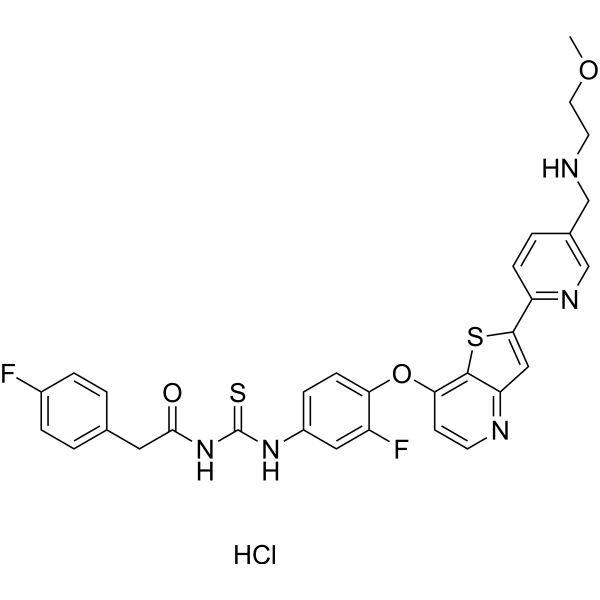
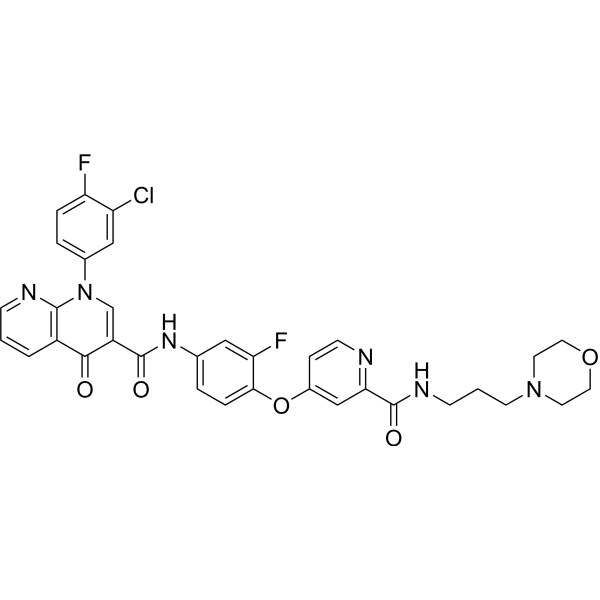
Zanzalintinib (XL092) is an orally active, ATP-competitive inhibitor of multiple receptor tyrosine kinases (RTKs) including MET, VEGFR2, AXL and MER, with IC50s in cell-based assays of 15 nM, 1.6 nM, 3.4 nM, 7.2 nM respectively. Zanzalintinib exhibits anti-tumor activity. Zanzalintinib has the potential for kinase-dependent diseases and conditions research .
Cabozantinib S-malate (XL184 S-malate) is a potent multiple receptor tyrosine kinases inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
Antitumor agent-111 (compound 46) is an c-Metkinase inhibitor (IC50=46 nM) with antitumor and antiproliferative activity. Antitumor agent-111 arrests cell cycle at G0/G1 phase, and induces apoptosis .
Unecritinib (TQ-B3101) is a potent EGFR tyrosine kinase inhibitor. Unecritinib shows anticancer activity. Unecritinib inhibits ALK, ROS1, and MET. Unecritinib has the potential for the research of solid tumor and relapsed or refractory ALK-positive anaplastic large cell lymphoma .
(rel)-Tivantinib is a potent and highly selective inhibitor of the receptor tyrosine kinase c-MET. (rel)-Tivantinib has two novel targets, GSK3α and GSK3β, which play an important role in the cellular mechanism of non-small cell lung cancer (NSCLC) .
CCT128930 hydrochloride is a potent and selective inhibitor of AKT (IC50=6 nM). CCT128930 hydrochloride has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). CCT128930 hydrochloride induces cell cycle arrest, DNA damage, and autophagy. Antitumor activity .
X-376 is a potent and highly specific ALK tyrosine kinase inhibitor (TKI) (IC50=0.61 nM). X-376 is a less potent inhibitor of MET (IC50=0.69 nM). X-376 displays potent anti-tumor activity .
Cabozantinib-d4 is deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
Altiratinib (DCC-2701) is a multi-targeted kinase inhibitor with IC50s of 2.7, 8, 9.2, 9.3, 0.85, 4.6, 0.83 nM for MET, TIE2, VEGFR2, FLT3, Trk1, Trk2, and Trk3 respectively.
Cabozantinib-d6 is the deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively[1][2][3].
RIPK3-IN-1 is a RIPK3 type II DFG-out inhibitor with an IC50 of 9.1 nM. RIPK3-IN-1 inhibits RIPK1 and RIPK2 with IC50s of 5.5 and >10 μM. RIPK3-IN-1 is also a c-Metkinase inhibitor with an IC50 of 1.1 μM .
Amuvatinib (MP470) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib (MP470) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
Amuvatinib hydrochloride (MP470 hydrochloride) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib hydrochloride (MP470 hydrochloride) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
(3S,4S)-Tivantinib is a potent and highly selective inhibitor of the receptor tyrosine kinase c-MET. (3S,4S)-Tivantinib has two novel targets, GSK3α and GSK3β, which play an important role in the cellular mechanism of non-small cell lung cancer (NSCLC) .
Poloppin is a potent, cell penetrant inhibitor of the mitotic Polo-like kinase (PLK) (IC50=26.9 μM) and prevents the protein-protein interaction via the Polo-box domain (PBD) (Kd= 29.5 μM). Poloppin selectively kills cells expressing mutant KRAS, enhancing death in mitosis. Poloppin is used for the study of KRAS-mutant cancers as single agents, or in combination with c-MET inhibitors .
Axl-IN-18 (compound 25c) is a potent and selective type II AXL inhibitor. Axl-IN-18 shows excellent AXL inhibitory activity (IC50=1.1 nM) and 343-fold selectivity over the highly homologous kinaseMET in biochemical assays (IC50=377 nM). Axl-IN-18 significantly inhibits AXL-driven cell proliferation, dose-dependently suppresses 4T1 cell migration and invasion, and induces apoptosis. Axl-IN-18 shows noticeable antitumor efficacy in a BaF3/TEL-AXL xenograft model .
Protein tyrosine kinases (PTKs) are key signaling molecules and important drug targets. Two classes of PTKs are present in cells: the transmembrane receptor PTKs (RTKs) and the nonreceptor PTKs. The RTK family includes the receptors for insulin and for many growth factors, such as EGFR, FGFR, PDGFR, VEGFR, and NGFR. RTKs are transmembrane glycoproteins that are activated by the binding of their ligands, and they transduce the extracellular signal to the cytoplasm by phosphorylating tyrosine residues on the receptors themselves (autophosphorylation) and on downstream signaling proteins. Their principal functions of PTKs involve the regulation of multicellular aspects of the organism. Cell to cell signals concerning growth, differentiation, adhesion, motility, and death are frequently transmitted through tyrosine kinases. In humans, tyrosine kinases have been demonstrated to play significant roles in the development of many disease states, including diabetes and cancers.
MCE designs a unique collection of 1055 compounds that act as a useful tool for PTKs-related drug screening and disease research.
Srctide is a biological active peptide. (This is a peptide substrate for many protein kinases, such as Blk, BTK, cKit, EPHA1, EPHB2, EPHB3, ERBB4, FAK, Flt3, IGF-1R, ITK, Lck, MET, MUSK, Ret, Src, TIE2, TrkB, VEGF-R1 (Flt-1) and VEGF-R2 (KDR).)
FAM-CSKtide is a biological active peptide. (This is a FAM labeled peptide substrate (Abs/Em = 494/521 nm) for C-terminal Src kinase (Csk) and many other kinases such as Axl, cKit, ERBB4, Fes, Flt3, IGF-1 R, MET, MUSK, PYK2, Ret, TIE2, TrkA, VEGF-R1 and VEGF-R2.)
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, Fc) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-hFc labeled tag. The total length of HGFR Protein, Human (HEK293, Fc) is 908 a.a., with molecular weight of 170 & 100-130 & 45-50 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, hFc) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-hFc labeled tag. The total length of HGFR Protein, Human (HEK293, hFc) is 908 a.a., with molecular weight of ~130.6 kDa.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (Biotinylated, HEK293, His-Avi) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-Avi, C-6*His labeled tag. The total length of HGFR Protein, Human (Biotinylated, HEK293, His-Avi) is 908 a.a., with molecular weight of 75-95 & 40-50 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, His) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-6*His labeled tag. The total length of HGFR Protein, Human (HEK293, His) is 908 a.a., with molecular weight of 130 & 80-95 & 40-50 kDa, respectively.
Cabozantinib-d6 is the deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively[1][2][3].
Cabozantinib-d4 is deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
Phospho-c-Met (Tyr1349) Antibody is a non-conjugated and Rabbit origined monoclonal antibody about 156 kDa, targeting to Phospho-c-Met (Tyr1349). It can be used for WB,IP assays with tag free, in the background of Human.
c-Met-IN-17 is a potent c-Metkinase inhibitor with an IC50 of 0.031 μM. c-Met-IN-17 can be used in anticancer research. . c-Met-IN-17 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Inquiry Online
Your information is safe with us. * Required Fields.